缺陷的降解酶加剧遗传性疾病
在罕见的遗传性存储疾病(例如Sandhoff病或Tay-Sachs综合征)中,由于缺少关键的酶,导致神经节苷脂积聚的代谢废物无法在神经细胞中妥善处置。对患者的后果是严重的,从行动受限到失明,智力下降和过早死亡。
波恩大学的研究人员报告说,他们发现了为什么这些神经节苷脂还会在患有其他储存疾病的患者中积聚并导致其恶化。他们的研究(“ 膜脂质及其降解化合物控制溶酶体腔小泡中的GM2分解代谢 ”)在线发表在《脂质研究》上。

神经节苷脂GM2的分解代谢取决于三种基因产物。这些基因中任何一个的突变都会导致不同类型的GM2神经节病(Tay-Sachs病,B1变异,Sandhoff病和AB变异),而GM2是主要的溶酶体贮藏化合物。GM2还是溶酶体贮积病(如A,B和C型Niemann-Pick病)中的次要贮藏化合物,分别主要贮藏SM和胆固醇。“在携带GM2的脂质体表面重构GM2分解代谢表明,将脂质掺入到携带GM2的膜中,例如胆固醇,SM,鞘氨醇和鞘氨醇,可抑制由GM2活化蛋白辅助的β-己糖胺酶A的GM2水解,而阴离子脂质,神经酰胺,脂肪酸,溶血磷脂酰胆碱和二酰基甘油刺激GM2分解代谢。
“此外,我们可以证明水解抑制脂质对GM2激活蛋白对膜结合脂质的增溶和动员也具有抑制作用,而刺激脂质则增强了脂质动员。”
在Tay-Sachs综合征和Sandhoff病中,神经细胞膜的成分无法适当降解,导致神经节苷脂GM2储存在溶酶体中。神经节苷脂脂质主要存在于神经系统的神经节细胞中。如果GM2降解酶Hex A缺失或受损(例如由于遗传缺陷),则会发生破坏性神经节苷脂储存。
在桑霍夫氏病中,降解酶Hex A和Hex B没有活性。与Tay-Sachs综合征一样,GM2的储存会导致神经细胞的破坏。患病的儿童在生命的头几个月中正常发育;后来,失明,行动受限和智力下降发生,并最终提前死亡。
波恩大学LIMES研究所高级教授Konrad Sandhoff博士说:“以前的治疗方法并未在这些神经变性神经节苷脂酶方面取得任何重大成功。” 酶替代疗法由于血脑屏障对这些物质的不可渗透性而失败。
桑德霍夫和他的研究小组已经破译了分子环境在溶酶体中对GM2成功降解的作用。在试管中,科学家重建了溶酶体中降解GM2的囊泡。通常,辅助蛋白GM2AP有助于捕获和释放位于囊泡壳上的GM2。然后可以将其与降解酶Hex A一起降解为无害的GM3。但是,当十六进制A的功能被阻止时,它会被存储,对神经细胞造成致命后果。
在试管中,科学家研究了抑制或改善GM2降解的影响因素。例如,囊泡越小,其表面带负电的越多,降解酶越容易接近GM2,“消化”功能越好。
另一方面,胆固醇和鞘磷脂的存在显着降低了GM2的降解。研究表明,这些脂质在尼曼-皮克病中的储存触发了溶酶体中额外的GM2积累,即使GM2降解酶Hex A完整且活跃,也显着加剧了C型临床表现。Sandhoff补充说:“降解酶的遗传疾病显然引发了一系列未知的后果性损害。”
在另一项研究中,研究小组表明,这种级联原理也适用于遗传性粘多糖多糖酶。在这些疾病中,一种储存物质硫酸软骨素通过抑制GM2降解,触发了神经细胞中神经节苷脂的额外储存。除了现有的身材矮小,面部特征粗糙和肝脏肿大以外,它还引起学习困难和惊吓反射,但是在动物模型中,可以通过抑制GM2的形成来缓解这种情况。
桑德霍夫说:“当前治疗方法的目的是防止针对这些遗传性存储疾病的GM2产生。”他补充说,目前市场上出售的药物仅能部分满足这一要求。“也许基因替代疗法已经在动物模型中获得成功,并将很快用于患者中,它将更加成功。”
推荐内容
-
红外发光纳米颗粒为研究人员提供了活体小鼠大脑内部的视图
斯坦福大学的化学家开发了一种新的深层组织成像技术,该技术可以看到活着的受试者的皮肤下方,以无与伦比的清晰度照亮埋藏的肿瘤。在9月30
-
水生藻类基因组包含古代陆地植物基因
一项新的全基因组关联荟萃分析已鉴定出44个与主要抑郁症有显着关联的基因组变异(基因座)。在这44个基因座中,有30个是新发现的,而先前的研
-
原发性和转移性乳腺癌可通过耗尽UBR5蛋白而发育不良
科学家已经发现了这种被称为UBR5的酶在推进原发性和转移性乳腺癌方面的新作用。转移是癌细胞从原发部位扩散到其他器官以形成新肿块的过程,
-
科学家们负责组织负责能源生产的细胞机制
Centro Nacional de Enfermedades Cardiovasculares Carlos III(CNIC)的科学家已经确定了活细胞能量产生的分子组织。发表在 自然
-
一种探测流体中DNA分子模式的方法
来自沙特阿拉伯的一个研究小组开发了一种新的分析方法,可以捕获均质液体中DNA等大分子的运动1。这种被称为晶格占据分析的新方法揭示了DNA
-
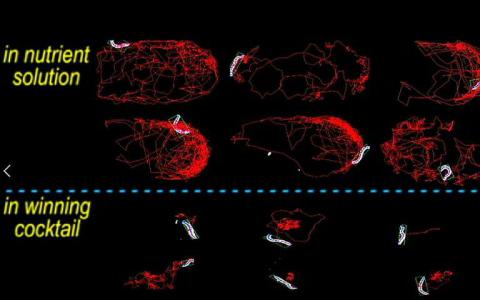
研究人员展示了结合药物控制寄生虫的工程方法
实验室视频讲述了这样一个故事:在营养液中自由游动的微小寄生虫是活跃的和可移动的;将同种蠕虫暴露于通过工程技术优化的四种药物的混合物
-
使用合适的植物可以减少室内污染并节省能源
工业化国家的人们在室内度过了80%以上的生活,越来越多地生活在气密的建筑物中。这些结构需要较少的能量用于加热,通风和空调,但如果颗粒
-
新的基因工程技术可以帮助设计 研究生物系统
一项新技术将帮助生物学家修补基因,无论目标是将细胞转变为生产药物的小工厂,修改作物以在有限的水中生长,还是研究基因对人类健康的...
-
法国葡萄园准备打破草甘膦成瘾
数十年来,法国葡萄园的引人入胜的土地已被世界上使用最广泛的除草剂所饱和,但葡萄种植者表示,当草甘膦不再是优质葡萄酒工艺的一部分...
-
多样性和移民提高了微生物群落的生产力
新的研究显示,自然选择很快将微生物的大熔炉变成了一个高效的社区。埃克塞特大学的科学家将十个不同的产甲烷群落(数百种微生物种群,主要