阅读长DNA序列的技术进步在理解灵长类动物进化 人类疾病方面具有分歧
阅读长DNA序列的技术进步在理解灵长类动物进化和人类疾病方面具有分歧。西部低地大猩猩的基因组现在已经被测序并以高质量水平组装,开始接近小鼠和人类基因组的基因组。基于较长序列读取的新测序技术允许首次发现缺失基因和缺失形式的遗传变异。这个集合为生物物种提供了新的生物学见解,这种生物物种仅次于黑猩猩,它与人类的进化接近。

华盛顿大学基因组科学教授埃文·艾希勒(Evan Eichler)领导的研究人员在4月1日出版的“ 科学”杂志上报道了为什么大猩猩和其他哺乳动物的先前基因组装配已经碎片化,不完整且可能具有误导性:
大规模并行测序技术在提高速度,提高准确度和降低基因组测序成本的同时,通常只产生称为“读数”的短序列序列。测序后,读数与基因组装配软件一起拼凑。该程序试图通过使用序列读数之间的重叠来重建原始基因组。不幸的是,人类和其他灵长类动物基因组中常见的长重复DNA的存在使装配软件混乱并导致其将基因组分解成非常小的片段。
“这种组装可以像瑞士奶酪一样,”艾希勒说,“缺口中缺少许多生物信息。” 他说,使用短读技术创建的原始出版的西部低地大猩猩基因组被分解成超过40万个。
“这些差距不是随机的,而是聚集在重复的位置,”他说。“如果遗传学家无法捕获这些重复序列并确定基因组中的结构差异,那么他们在理解基因组织和比较物种内部和物种之间的遗传变异方面存在问题。”
他的团队包括威斯康星大学生物信息学专家David Gordon和John Huddleston,以及博士后研究员Mark Chaisson,Chris Hill和Zev Kronenberg。研究小组在芝加哥林肯公园动物园的一只雌性西部低地大猩猩的血液样本中分析了DNA。
研究人员使用单分子,实时(SMRT)测序技术,装配工具Falcon和QUIVER,以及其他技术来产生长序列读数。这些是最流行的序列技术的一百多倍。长读取允许它们在组装期间穿过大猩猩基因组的大部分重复区域。
结果是一个新的大猩猩基因组装配体更大,并且具有更少的碎片。现在只有1,800件,而不是400,000件。基因组片段的平均大小为800倍,原始组件中所有间隙的约90%关闭。
研究人员观察到,这种额外的测序信息大大改善了该种大猩猩的基因注释。它还导致发现了数千个蛋白质和肽编码区段以及作为第一个基因组组装的一部分而遗漏的新的调控元件。基因如何被控制,甚至某些基因调控元素的丢失或破坏的差异,可以解释为什么人类祖先的进化与他们的大猿亲属如此不同。
科学家还发现了数以万计的新结构变异,例如DNA的缺失或插入,这些变异可能比之前编目的较小的单碱基对差异更重要。(碱基对是连接到DNA梯子上的梯级的两种化学物质)
“我研究人类和大猿基因组的动机,”艾希勒说,“是试图了解是什么让我们成为一个物种。我希望看到所有伟大的猿类基因组重新做,包括黑猩猩和猩猩,以全面了解区分人类和类人猿的遗传变异。我相信遗传变异远远超过我们之前的想法。第一步就是找到它。“
在研究人员看到人类和大猩猩之间有趣的差异的领域中,有与感官知觉,角蛋白(皮肤蛋白)产生,胰岛素调节,免疫,繁殖和细胞信号传导相关的基因。
新的基因组装配也为低地大猩猩的进化历史提供了新的线索。之前的研究表明,大猩猩种群在不久的将来经历了一个瓶颈,但用新基因组进行的分析表明,瓶颈比以前认为的更严重。
大猩猩基因组内的遗传变异模式可以提供疾病,气候变化和人类活动如何影响低地大猩猩种群的证据。
“我认为带回家的信息,”艾希勒说,“新的基因组技术和装配将我们带回了10年前我们应该去过的地方。”
“测序技术和计算生物学,”Eichler和他的团队在他们的论文中写道,“现在已经进入了各个实验室可以产生高质量哺乳动物基因组的阶段。这种能力有望彻底改变我们对基因组进化和物种生物学的理解。 “。
艾希勒补充说,这些进展也可能对人类疾病的遗传基础研究做出巨大贡献,特别是如果以这种方式对更多的人类基因组进行测序。
“作为医学研究人员,如果我们只依赖于短读序列,那么我们的装甲就会出现问题。大猩猩和其他人类基因组的工作清楚地表明,用短序列读取方法无法理解大片的遗传变异。长读序列使我们能够获得以前难以接近,无法进入的新水平的遗传变异,“他说。
然而,他补充道,“流行价为80,000美元,使用长读数对人类基因组进行临床测序的价格尚未正确。鉴于几年的成本降低和技术的进一步发展,我愿意打赌这个我们将在未来对人类基因组进行测序以发现引起疾病的突变。“
推荐内容
-
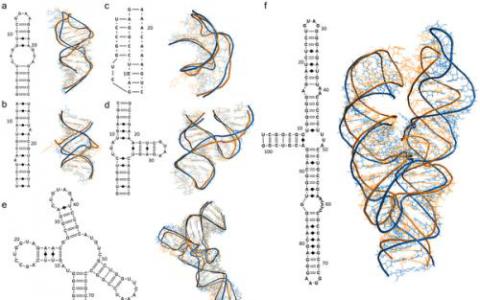
三维治疗疾病-超级计算机有助于解码RNA结构
治愈癌症,艾滋病毒和其他顽固疾病已经回避了几代人最聪明的头脑。但随着超级计算机 - 可以计算,分析和可视化极大量数据的计算系统 -
-
Lonza推出新的基础和饲料培养基以优化蛋白质生产
龙沙生物治疗媒体全球产品经理Nicole Wellens:生物制药研究人员需要使用高质量的培养基,这些培养基可以提供恒定流量的健康且有活力的细
-
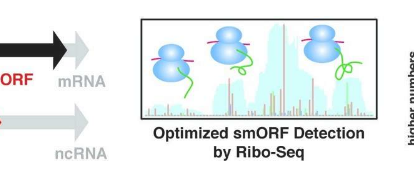
寻找最小的基因可能会产生巨大的收益
尽管科学家们知道大约25,000个编码重要生物学蛋白质的基因,但隐藏在我们DNA中的其他较小基因可能同样重要。但是事实证明,这些微小的遗传
-
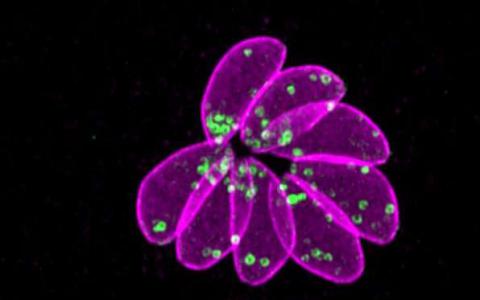
当研究人员取下手制动器时寄生虫会快速退出
墨尔本研究人员已经发现了一种方法来阻止导致弓形虫病的寄生虫进入细胞,从而使寄生虫丧失其生长所必需的关键因子。该研究结果是接近疫...
-
寨卡病毒感染基因模式的菌株差异
科学家已经揭示了非洲寨卡病毒和亚洲寨卡病毒如何感染神经祖细胞之间的分子差异。这些结果可以提供对Zika病毒最近作为全球健康紧急事件而出
-
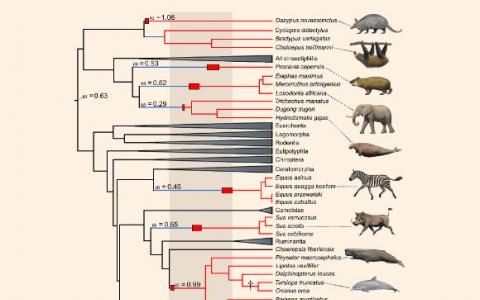
单基因与多个哺乳动物谱系的进化兴起有关
在古代,独立inactivationsUCP1基因,其编码解偶联蛋白1,或UCP1(必要的热产生棕色脂肪),为驾驶的近一半胎盘的哺乳动物订单进化的主要力量
-
新的突变珊瑚共生藻类能够关闭共生
日本的研究人员已经确定了第一个自发的突变珊瑚共生藻,它不与宿主保持共生关系。他们设计了一种系统,其中营养素的简单添加或消耗可以...
-
基因疗法可恢复先天性耳聋
由法国科学家领导的一个国际研究小组利用基因疗法来恢复成年小鼠的听力,这些小鼠患有DFNB9耳聋,这是最常见的遗传性或先天性耳聋之一。除
-
两栖皮肤细菌在寒冷 多变的环境中更加多样化
两栖动物是致命的皮肤病流行病的受害者。在第一项全球范围的研究中,包括史密森热带研究所(STRI)在内的31所大学和研究中心的研究人员收集了
-
盐生长的浮游动物生长太慢 不能阻止盐诱导的藻类繁殖
淡水食物链底部的小动物可以迅速适应盐污染 - 来自冬季道路除冰,农业和采矿等来源 - 但需要付出代价。在专门讨论淡水盐污染的皇家学会