神经网络检测单细胞RNA-Seq
卡内基梅隆大学(Carnegie Mellon University, CMU)的计算机科学家报告称,神经网络和监督机器学习技术可以有效地表征使用单细胞rna测序(scRNA-seq)进行研究的细胞。研究小组称,这一发现有助于研究人员识别新的细胞亚型,并区分健康细胞和患病细胞。

这种新的自动化方法不依赖于标记基因,而标记基因并不是对所有细胞类型都可用,而是分析所有的scRNA-seq数据,从而选择能够区分不同细胞的参数。来自CMU计算生物系的研究人员指出,这使得对所有细胞类型的分析成为可能,并为这些细胞的比较分析提供了一种方法。他们的研究(“单细胞RNA-seq数据比较分析的web服务器”)发表在《自然通讯》杂志上。他们说,名为scQuery的web服务器使所有研究人员都可以使用这种方法。
单细胞RNA-Seq (scRNA-seq)研究了异质环境中的数千个细胞。目前描述细胞特征的方法是先进行无监督分析,然后使用一组已知的标记基因进行赋值。这种方法仅限于少数特征良好的细胞类型。我们开发了一个自动化的管道来下载、处理和注释公开可用的scRNA-seq数据集,以支持大规模的监督描述,”研究人员写道。
“我们扩展了监督神经网络,以获得高效和准确的表示scRNA-seq数据。我们使用我们的管道分析了来自超过500个不同研究的数据,其中有超过300种独特的细胞类型,并表明监督方法在细胞类型识别方面优于非监督方法。一项案例研究强调了这些方法在比较健康和患病小鼠细胞类型分布方面的有效性。最后,我们介绍了scQuery,这是一个web服务器,它使用我们的神经网络和快速匹配方法来确定细胞类型、关键基因等。
在过去的五年中,单细胞测序已经成为细胞研究者的主要工具。在过去,科学家只能通过处理成批的细胞来获得DNA或RNA序列信息,提供的结果只能反映细胞的平均值。相比之下,一次分析一个细胞可以使研究人员识别出细胞的亚型,或者看到健康细胞与患病细胞的区别,或者年轻细胞与衰老细胞的区别。
这种类型的排序将支持美国国立卫生研究院的新人类生物分子图谱计划(HuBMAP),这是建立一个3 d地图显示的人体组织细胞水平不同,根据bar - joseph Ziv,博士,教授,计算生物学和机器学习和今天的论文的作者之一,领导CMU-based中心贡献计算工具项目。
计算生物学博士生阿米尔·阿拉维(Amir Alavi)和博士后研究员马修·鲁法洛(Matthew Ruffalo)博士共同撰写了这篇论文。阿拉维说,“随着每个实验产生数十万个数据点,这正成为一个大数据问题。传统的分析方法不足以应对如此大规模的数据。”
Drs。Alavi、Ruffalo和他们的同事开发了一个自动化的管道,试图下载所有可用于mico的公共scRNA-seq数据——识别每个细胞中表达的基因和蛋白质,这些数据来自最大的数据存储库,包括NIH的基因表达总汇(Gene Expression Omnibus, GEO)。然后,这些细胞被按类型进行标记,并通过神经网络进行处理。神经网络是一种模拟人类大脑的计算机系统。通过比较所有的细胞,神经网络识别出使每个细胞不同的参数。
研究人员使用小鼠类似阿尔茨海默病的scRNA-seq数据测试了这个模型。分析显示,健康细胞和患病细胞的脑细胞水平相似,而患病细胞中包含了更多的免疫细胞,如巨噬细胞,它们是在对疾病的反应中产生的。
研究人员使用他们的管道和方法创建了scQuery,这是一个web服务器,可以加速对新的scRNA-seq数据的比较分析。一旦研究人员向服务器提交一个单细胞实验,该小组的神经网络和匹配方法就可以快速识别相关的细胞亚型,并识别早期对类似细胞的研究。
推荐内容
-
突破性的研究显示植物如何感知世界
植物缺乏眼睛和耳朵,但他们仍然可以看到,听到,闻到和应对环境线索和dangers-especially致命的病原体。他们这么做的帮助下数以百计的膜蛋白,
-
4位生殖中心护士,讲述与试管婴儿患者间的温情故事
4位生殖中心护士,讲述与试管婴儿患者间的温情故事 原标题:4位生殖中心护士,讲述与试管婴儿患者间的温情
-
7月30日辽宁沈阳疫情最新实时消息公布 沈阳提醒确诊病例所在街
温馨提示:疫情期间规范佩戴口罩,注意乘坐公共交通工具、进入人员较为密集或通风不良的场所、到医院就诊,以及出现呼吸道症状时应自觉...
-
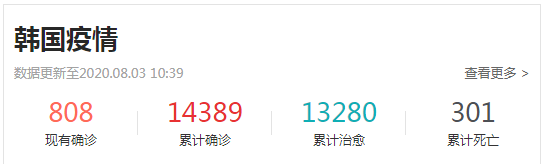
韩国为防病毒用微波炉烤钱 破损钱币面积为3/4以上时可换新币
目前韩国受新冠肺炎影响,韩国民众为了预防新冠病毒传播,已经开始被迫采取了各种各样的方法。因此,今天就有一则关于韩国为防病毒用微波炉
-
8月5日张家界疫情最新实时数据公布 张家界昨日新增新型冠状病
你知道张家界目前疫情什么情况吗?张家界疫情有几人确诊了?据消息显示,湖南8月4日新增本土确诊9例:株洲1例,张家界8例。2021年8月4日0--24
-
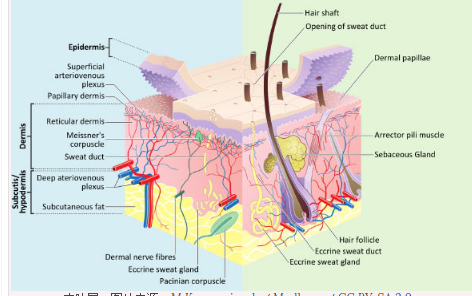
皮肤细胞发现可能导致脱发 头发灰白的可能治疗
德克萨斯大学西南医学中心的一组科学家已经鉴定出直接产生头发的细胞,以及导致头发变白的机制。这项研究发表在《基因与发展》杂志上。Haro
-
新冠疫苗最新消息 中国新冠疫苗生产线调试安装
新冠疫苗一般指新型冠状病毒疫苗。新型冠状病毒疫苗,是针对新型冠状病毒的疫苗。据最新消息显示,刚刚就有一则关于中国新冠疫苗生产线...
-
广州婷婷是谁个人资料简介及照片曝光 广州婷婷服饰辱骂河南人
一个叫婷婷的把河南人全都罪了,好多人都在网上找她。究竟是怎么回事呢?据消息显示,这几天河南的灾情牵动着全国人民的心,然而这时爆出...
-
全世界第一针新冠疫苗打在武汉 “人民英雄”陈薇院士透露抗疫细节
【导读】新冠疫苗的作用是通过在人体内接种灭活病毒,来抵抗外界病毒的侵入,这是一种比较有效的抵抗病毒的方法。据最新消息显示,全世...
-
数十名外卖小哥11秒抬车救人 网友:“正能量的代言”
【 数十名外卖小哥11秒抬车救人 】刚刚一则关于骑车女子被压车底数十名外卖小哥11秒抬车救人的消息登上了热搜榜!据最新消息显示,昨天杭