AdvaMed赢得了一些 在FDA关于管理不确定性的指导中失去了一些
该指南的存在是因为FDA在决定是否授权设备时必须管理不确定性,无论是通过上市前批准(PMA),De Novo分类还是人道主义设备豁免(HDE)。为确保患者及时获得新设备,FDA会权衡其可能带来的好处和风险,而不是要求公司预先生成详尽的数据。

对突破性设备尤其如此,这些设备在治疗上非常重要,FDA可能愿意接受更多的不确定性。为了应对这种不确定性,FDA的指南草案表示公司可能必须收集上市后数据并在咨询委员会会议上进行讨论。该提案的部分内容遇到了行业的阻力。
“我们不相信咨询委员会会议应该是原子能机构的默认机制,”AdvaMed在其反馈中写道。“在某些情况下可能需要咨询委员会会议,但在所有情况下,它们都需要大量的时间和资源。赞助商花费数千小时和数十万美元(或更多)来筹备这些会议并不罕见。“
FDA在反馈后软化了其语言,但咨询委员会会议仍然是其计划的核心。在草案中,FDA表示它“打算”举行咨询委员会会议。在最终确定测试时,FDA将“打算”改为“普遍预期”。
这一变化是部分解决AdvaMed提出的问题的几项修订之一。该贸易组成功地呼吁FDA扩大其针对小患者群体开发设备的方法,并指出它将根据具体标准逐案做出决策。
然而,FDA在很大程度上抵制了在整个文本中取代“适当”这一术语的要求,AdvaMed称这种术语含糊不清,具有“合理性”,该组织称这是FDA及其利益相关者熟悉的法律和法规中的一个完善的术语。 。该机构似乎也主要忽视了关于常规PMA,De Novos和HDE的更多信息的呼吁,因为重点关注突破性设备和AdvaMed呼吁的小患者人群。
FDA还收到了消费者权益组织“公共利益科学中心”的反馈,该组织提出了一系列不同的担忧。正如CSPI所看到的那样,指南中引用的例子提供了“明确的降低监管标准的垫脚石”,这将使患者面临风险。
“毫无疑问,公司将抓住这些情景,就特定设备应如何接受比机构可能设想的更大程度的不确定性而进行争论,从而将数据需求转移到上市后期间,从而减少上市前样本量,“CSPI写道。
FDA在最终确定文件时没有对这些例子做出重大修改。
推荐内容
-
6月12日南宁疫情最新数据公布 南宁市发现首例输入性新冠病毒印
【导读】疫情期间,小编还是那句话,虽然如今全国大部分地区都很安全,但对抗疫情的路还很长,所以别掉以轻心!印度新冠肺炎疫情持续暴发以
-
土壤益生菌承诺更大更健康的作物但有一个缺点
超过一半的世界工厂所产生的能量摄入量来自于刚刚三种作物:水稻,小麦和玉米。与大多数陆地植物一样,这些作物与一种称为丛枝菌根真菌...
-
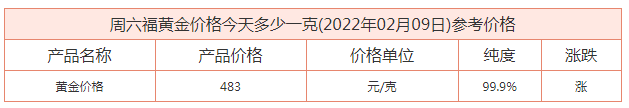
周六福2022金价今日价格多少钱一克?香港周六福铂金今日价格多少钱
今日黄金价格及今日金价查询(黄金首饰价格多少一克,今日黄金首饰价格走势,黄金饰品价格多少一克查询),想必这两天大家最关注的就是金价一事
-
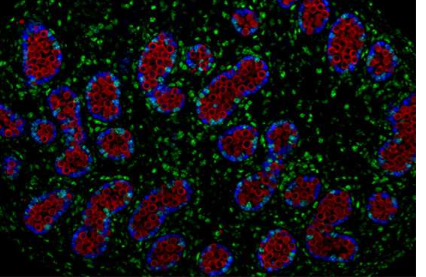
原始生殖细胞如何在胚胎性腺中开始精子和卵子的产生
在哺乳动物胚胎发育的早期,很早在生物体的最终形态形成之前,就将其细胞的宝贵子集留作后代以用于后代的繁殖。这项任务赋予该细胞子集...
-
12月12日辽宁大连疫情最新数据公布 辽宁昨日新增1例境外输入确
【 辽宁新增1例境外输入确诊病例 】12月11日0时至24时,辽宁省新增1例境外输入新冠肺炎确诊病例,为大连市报告。当日治愈出院境外输入无症
-
10月28日威宁自治县疫情最新消息公布 威宁自治县有序开展
威宁自治县有序开展新冠疫苗加强针接种。据消息显示,近日,在威宁自治县疾病预防控制中心疫苗接种点,群众有序前来接种加强针。昨天菜...
-
11月18日香港迪士尼乐园疫情最新消息公布 香港迪士尼乐园将于1
香港迪士尼乐园将于11月19日重新开放。据香港迪士尼乐园官网11月18日消息,香港迪士尼乐园将于11月19日重新开放。
-
李云迪绯闻女友是谁结婚了吗?李云迪事件女主是陈思卉吗照片视频
李云迪绯闻女友被倒追结婚了吗 李云迪事件女主陈某卉照片视频炫富真的吗?而对于这两天闹得沸沸扬扬的李云迪事件,今天的你是否也在关注着
-
微信小老虎后缀怎么弄方法2022 微信小老虎设置方法最新步骤
关于微信小老虎后缀怎么弄?微信小老虎后缀设置方法的这个话题,相信小伙伴们是非常有兴趣了解的,因为这个话题是目前网络上非常火热的,既
-
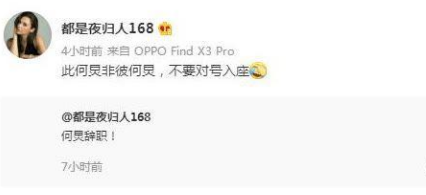
快乐大本营停播了吗节目停播的原因是什么?何炅辞职是真的吗为什
近日一则关于《快乐大本营》停播?节目单已更换为电视剧的消息引起了网友们的关注。想必大家对于快乐大本营停播了吗一事是非常关注的。具体