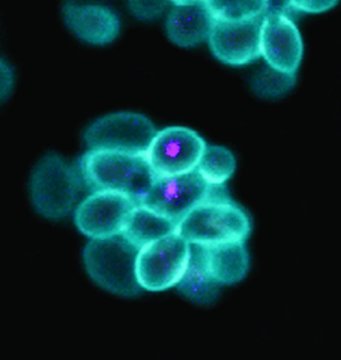
改进单细胞RNA测序数据可视化的新方法
就像从针孔相机到宝丽来,对流行的生物信息学数据可视化方法的公式的重要数学更新将允许研究人员开发单细胞基因表达的快照,不仅快几倍,而且分辨率更高。发表于自然方法,耶鲁数学家的这项创新将把百万点单细胞RNA测序(scRNA-seq)数据集的渲染时间从三个多小时缩短到仅十五分钟。
科学家表示,现有的十年前方法,即分布式随机邻域嵌入(t-SNE),非常适合用于表示在单一细胞水平上收集的RNA测序数据中的模式,scRNA-seq数据,二维。“在这种情况下,t-SNE'通过它们表达的基因组织细胞,并用于发现新的细胞类型和细胞状态,”首席作者,耶鲁大学博士生导师George Linderman说。专攻应用数学的学生。
根据计算标准,t-SNE非常慢。因此,在应用t-SNE之前,研究人员经常对其scRNA-seq数据集进行“下采样” - 从初始样本中采集较小的样本。然而,下采样是一个糟糕的妥协,因为它使得t-SNE不太可能捕获稀有细胞群,这通常是研究人员最想要识别的。
30多年前,另一支耶鲁大学数学家团队开发了快速多极方法(FMM),这是一种革命性的数值技术,可加速计算n体问题中的远程力。本研究的研究人员认识到,FMM背后的原理也可以应用于非线性降维问题,如t-SNE和加速t-SNE,直到它获得新名称:FIt-SNE,或基于快速插值的t -SNE。
“使用我们的方法,研究人员不仅可以更快地分析单细胞RNA测序数据,而且还可以用来表征在t-SNE之前对数据进行二次采样时无法检测到的稀有细胞亚群,”Yuval Kluger说道。作者和耶鲁病理学教授。此外,该团队对其FIt-SNE结果使用热图式可视化,这使研究人员可以轻松地同时查看数百个基因在单个细胞水平上的表达模式。
研究人员表示,2019年t-SNE获得“FIt”的新年不会更好。2018年12月,“科学杂志”命名为逐个细胞跟踪胚胎发育 - 如果没有基于scRNA-seq数据的可视化 - 年度突破,则无法实现。研究人员表示,FIt-SNE将加速这一发育生物学领域以及神经科学和癌症研究领域的进一步工作,其中单细胞测序已成为绘制大脑和理解肿瘤的宝贵工具。
推荐内容
-
合成微生物使科学家们能够研究古老的进化神秘
斯克里普斯研究中心的科学家及其合作者创造了微生物,这些微生物可能概括了数十亿年前被认为生存过的生物的关键特征,使他们能够探索生...
-
通过扩大专科护理可以改善慢性肾病的结果
根据兰德公司的一项新分析,在需要透析治疗之前为肾脏衰竭的患者提供专业的医疗护理和协调,每年可为美国医疗保健系统节省超过10亿美元...
-
团队发现使用2-D材料控制光相位的新方法
随着研究人员寻求方法来满足对信息处理和通信的不断增长的需求,在纳米级或纳米光子学上的光学操纵已成为一个关键的研究领域。控制和操...
-
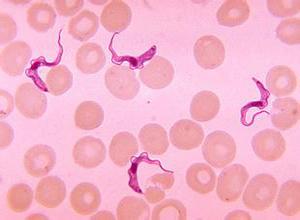
发现一种潜在的治疗靶点来对抗锥虫
Yaser Hashem在CNRS的实验室建筑和研究中心的团队发现了一种新的潜在治疗靶点 - 位于核糖体中 - 用于对抗锥虫寄生虫。使用低温电子显
-
高血压药物增加阿尔茨海默氏病患者的海马血流量
脑血管变化,包括脑血流量减少,发生在阿尔茨海默氏病的发展早期,可能会加速疾病的进展。在本周在线发表于《高血压》杂志上的一项研究...
-
9月14日泉州鲤城区疫情最新消息公布 泉州鲤城区公布一阳性潘
温馨提示:一旦出现发热、干咳、乏力、鼻塞、流涕、咽痛、嗅觉味觉减退、结膜炎、肌痛和腹泻等症状,应及时按规范程序就诊,并主动告知1...
-
新的临床试验寻求治疗犬癌症可能提供人类癌症的线索
目前正在通过塔夫茨大学康明斯兽医医疗中心新成立的临床试验办公室开展两项关于犬致命癌症的研究。患有自发性骨肉瘤的狗以及患有肥大细...
-
受骨骼和软骨启发的膜可从盐水中高效发电
受活生物体组织中的膜的启发,科学家将用于凯夫拉尔的芳族聚酰胺纳米纤维与氮化硼结合在一起,制成了一种膜,用于收集既像骨头一样坚固...
-
研究暗示了鳞状恐龙腿如何能获得鸟状羽毛
犹他大学的科学家们确定了两个基因,这些基因使一些鸽子品种形成羽毛状的脚,称为套管,而其他基因则缩小了脚。相同或相似的基因可以解...
-
抗生素抗性基因增加
在世界各地,抗生素的使用和抗药性正在增加,而新抗生素的发现几乎停止。在密歇根州立大学进行的一项新研究中,发表在最新一期的mBio杂志上